The proteome doesn't have a coverage gap – it has a coverage bias
We can now measure thousands of proteins in a drop of blood.
The ones we still can't measure aren't a random remainder – and that bias shapes what medicine can find.
Proteins do almost everything in the body. They carry oxygen, fight infections, switch genes on and off, and pass signals between cells. The full set a human body makes – its proteome – is what most of modern medicine is ultimately trying to read.
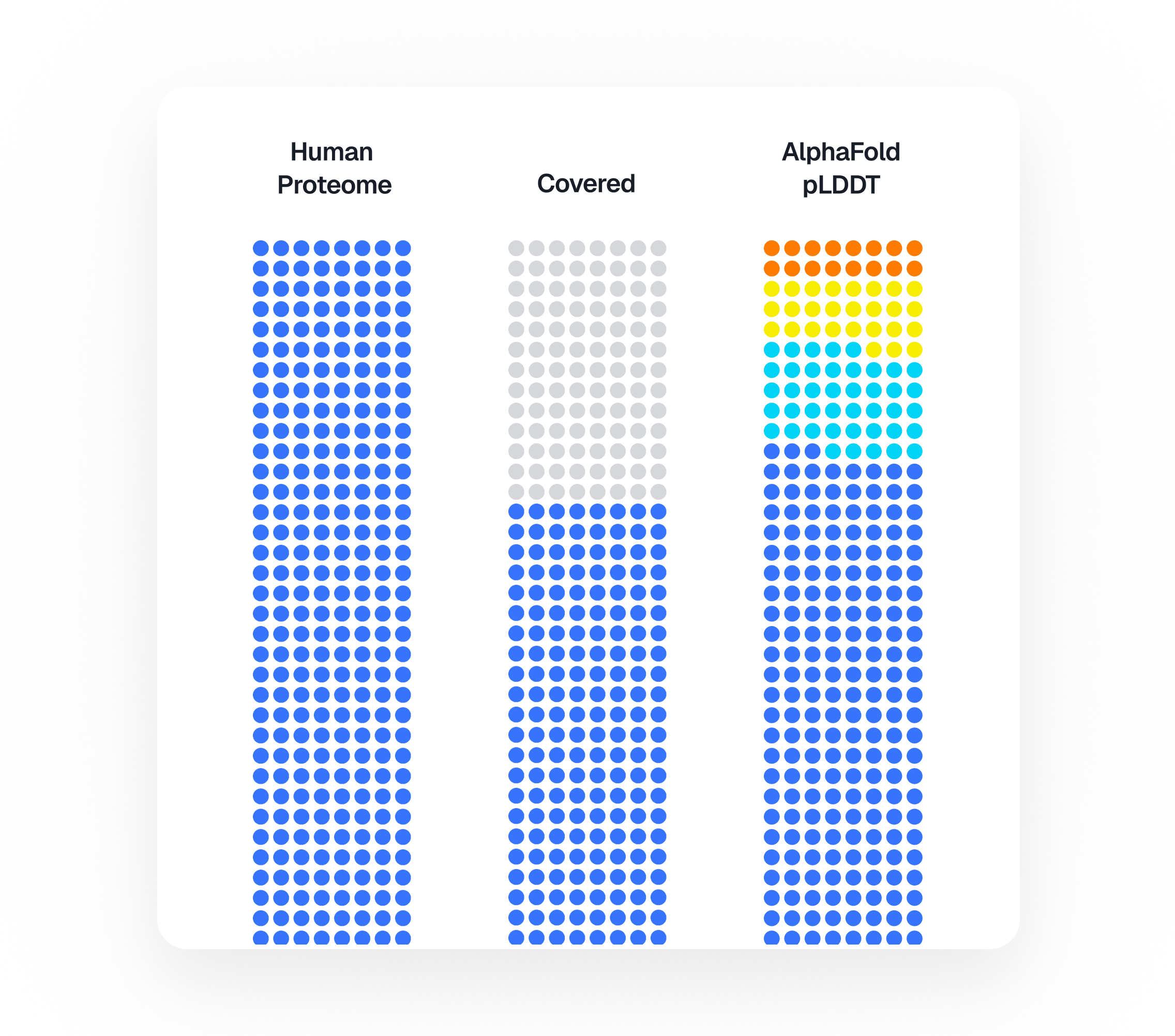
The count of proteins scientists can measure in a sample of blood has been climbing for years: from hundreds, to thousands, to around ten thousand today, roughly half the body’s protein-coding genes1. Put like that, the job sounds nearly done. Keep improving the technology, add proteins, and eventually the list will be complete.
It doesn't work that way. The proteins we still cannot measure are not a random leftover waiting to be picked up on the next pass. They are a specific kind of protein, absent for particular reasons. And several of those reasons point straight at the biology we most want to understand.
How far we've come
The progress is real. A handful of platforms can now measure thousands of proteins at once from a tiny sample2. The largest effort of its kind, the UK Biobank Pharma Proteomics Project, is measuring about 5400 proteins from six hundred thousand samples, cross-referenced against each donor's genetics and medical record to surface early signs of disease and find new targets for drugs. Its 2023 pilot covered about 2900 proteins in 54000 people, the jump gives a sense of the pace. The field's two rival platforms – Olink, built on antibodies3, and SomaLogic's (now part of Illumina) aptamer-based SomaScan – now cover more than half of the human proteome between them4,5.
But "measuring a protein" turns out to mean several different things, and they are easy to confuse. Knowing a protein exists is one thing. Catching it in a single sample is another. Reading it reliably across hundreds of thousands of people is harder still. Hardest of all is telling apart the several forms one protein can fold or split into6. A protein can be easy to measure in a piece of tissue and invisible in blood, or show up in a careful one-off experiment and then fail in a pipeline that runs thousands of samples a day. So a headline number like "ten thousand proteins" hides a great deal of fine print.
The leftover proteins have something in common
Line up the proteins that today's big blood tests miss, and a pattern appears. They tend to share awkward properties.
Many work inside cells and seldom leak into blood, so no assay, however sensitive, will find them there. Others hold their working shape only while they are embedded in the cell's outer membrane, pull them out, and the shape you need to recognize collapses. Some come in nearly identical copies – families that differ by only a few building blocks – so a binder that grabs one grabs them all. And some have no fixed shape at all until they clamp onto a partner molecule.
You can put faces to this. The proteins that switch genes on and off are a large and important group, and they mostly stay inside the cell and look very much alike, which is the exact combination that defeats current tests. The receptors that let a cell sense its surroundings run straight through the membrane and hold their shape only there. These are not obscure molecules. They are central to how the body works, and they are precisely the ones left out.
Consider two of them.
Notch2 is a receptor embedded in the outer membrane of a cell, where it passes signals inward. When it fires it breaks apart: one part is shed outside the cell, another is released inward to switch genes on. Certain cancers grow by keeping this switch stuck on, so a test that reported whether Notch2 is actively signaling – not just how much of it is present – would help flag those tumors and show whether a Notch-blocking drug is working. A single abundance number can't do that. What floats in blood as "Notch2" is really a mix of fragments, averaged into one figure that says nothing about whether the receptor is doing its job.The way out is a binder raised against the cleaved, active fragment alone – the piece released when the receptor fires – so the readout tracks signaling rather than bulk. Assays that pick a protein's forms apart like that barely exist yet, and building them is much of the unfinished work.
Sox2 fails a blood test for the opposite reason. It works in the nucleus, gripping DNA, helping steer what a cell becomes; too much of it marks squamous cancers of the lung, esophagus, and head and neck, so a reliable blood readout would help catch or track them. But Sox2 was never meant to be in circulation. When it shows up there, it usually just means cells have burst. Its real work happens bound to DNA and partner proteins, and the stray, unbound copy a blood test might snag is not the copy that matters.
Neither protein is obscure. Both would be worth tracking. And both are the kind routine blood work skips – not for lack of importance, but because of how measurement itself is built.
So "the proteins we can't measure yet" isn't one problem with one fix. For some, we simply haven't built the right recognizing molecule. For others, we are looking in the wrong sample. For others still, the protein takes several forms and the test cannot tell them apart.
The bias is built in, one reasonable step at a time
None of this comes from carelessness. It is the sum of sensible engineering choices, each of which made large-scale measurement possible in the first place. But each choice also quietly favors some proteins over others.
It begins with the sample. Blood is convenient, but it is not a summary of the whole body. It is one fluid, in which the most common proteins outnumber the rarest by a factor of ten billion or more. A protein that is central to a disease can be a poor thing to look for in blood, simply because it stays inside a rare type of cell and never really gets out. When that is the case, the fix is not a more sensitive instrument. It is a different sample – which is why the same technologies are now being extended beyond blood into spinal fluid and tissue.
Next, the target has to be turned into something you can test against: a purified version of the protein in a tube. If that version does not match the shape the protein has in the living body, the recognizing molecule you find will be precise about the tube and useless in a patient.
The recognizing molecule itself, usually an aptamer or antibody, has to be discovered from a limited library. When nothing in the library fits, you can't tell whether the protein is genuinely intractable or you just didn't have the right net. That feeds a loop: proteins with a good binder get measured more, studied more, funded more, which yields still more binders for the same proteins. The roster of well-covered proteins tracks importance, yes, but it also tracks which proteins happened to draw a good binder early.
And the assay has to hold together as a whole. Thousands of binders share one tube, and one that performs beautifully in isolation can misbehave in that crowd; a slightly worse binder that plays nicely with the rest is often the better choice. A binder that works alone still has to survive the company of thousands of others in the same tube.
A better way to pose the question
The newer part of this starts before the bench at all. For most of the field's history, finding a binder meant brute force: make the protein, throw a big library at it, keep whatever sticks. That still works, and it's still most of what happens. What's changed is how much can be worked out ahead of time.
Structure-prediction models – AlphaFold 2, released in 2021, and the tools that followed7 – can now show on a screen what a protein looks like, which parts are exposed, how it diverges from its near-twins, and whether the patch you want to grab is even reachable. A blind search becomes a pointed question: does this binder catch this patch of this protein, in the shape it actually holds?
And prediction is shading into design. The newer methods don't stop at reading a structure; they invent a molecule to fit it. RFdiffusion and ProteinMPNN, design new proteins to fit a chosen target: one invents a shape that would grip it, the other works out a sequence that folds into that shape. Companies like Absci use AI to raise antibodies even where no example binder exists. Others, such as Isomorphic Labs and the aptamer-design company Xelari, are constructing design engines on the same footing. The pitch is that instead of screening only what nature and prior effort happen to leave lying around, you could design a binder for a protein nobody has ever pinned down.
It's easy to oversell. A predicted structure or a designed molecule is a hypothesis until an experiment says otherwise, and the work stays a loop: predict, build, test, revise. The models don't replace the experiments. They decide which ones are worth running first.
What is actually at stake
The question was never only how many proteins we can measure. Counting up to ten thousand, and then to twenty, tells you how far a set of technologies has come. It does not tell you what you still cannot see, or why.
The sharper question is which proteins are missing, what they share, and at which step each one drops out. Framed that way, the missing half stops reading as blank space and starts reading as a worklist sorted by cause. Some proteins need a different sample. Some need a binder that doesn't yet exist. Some need an assay that can finally separate their forms.
This isn't housekeeping. The hardest proteins to measure are, disproportionately, the ones that run the nervous system, regulate genes, and study the cell surfaces where most drugs act. While they stay off the list, the big studies combing blood for the roots of disease are combing a skewed sample of biology – sharp where measurement is cheap, blank where it's hard – and every biomarker they surface, every drug target they float to the top, is drawn from that skew.
One thing has genuinely changed: the missing proteins are no longer a black box. For many we can now say why they resist, and for the first time we can try to design the specific molecules that would bring them in. That won't make the hard proteins easy. The next stretch of progress will come from deciding, deliberately, to go after what has been left out – and, for the first time, knowing where to aim.
References
- Kraemer, S. et al. “Crossing the Halfway Point: Aptamer-Based, Highly Multiplexed Assay for the Assessment of the Proteome.” Journal of Proteome Research 23 (2024). DOI: 10.1021/acs.jproteome.4c00411.
- Kirsher, D. Y. et al. “Current landscape of plasma proteomics from technical innovations to biological insights and biomarker discovery.” Communications Chemistry 8, 279 (2025). DOI: 10.1038/s42004-025-01665-1.
- Assarsson, E. et al. “Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability.” PLoS ONE 9, e95192 (2014). DOI: 10.1371/journal.pone.0095192.
- Katz, D. H. et al. “Proteomic profiling platforms head to head: Leveraging genetics and clinical traits to compare aptamer- and antibody-based methods.” Science Advances 8, eabm5164 (2022). DOI: 10.1126/sciadv.abm5164.
- Pietzner, M. et al. “Synergistic insights into human health from aptamer- and antibody-based proteomic profiling.” Nature Communications 12, 6822 (2021). DOI: 10.1038/s41467-021-27164-0.
- Smith, L. M., Kelleher, N. L. and Marshall, A. G. “Proteoform: a single term describing protein complexity.” Nature Methods 10 (2013). DOI: 10.1038/nmeth.2369.
- Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). DOI: 10.1038/s41586-021-03819-2.
© 2026 Xelari Inc. All rights reserved.